The Diagnosis is in the Hands
Brandon Janssen1, MD, Diana Gomez Manjarres2, MD, and Divya C. Patel2, DO
1Department of Medicine; University of Florida
2Division of Pulmonary, Critical Care and Sleep Medicine; University of Florida
Case
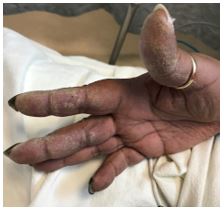
A 48-year-old woman with a reported history of sarcoidosis presented to the emergency department with worsening dyspnea on exertion, generalized fatigue, and cough productive of yellow sputum. She also reports chronic joint pain and the findings on her hands as shown.
The review of systems was negative for fevers, chills, night sweats, unintentional changes in weight, myalgias, muscle weakness, gastroesophageal reflux, and Raynaud’s phenomenon. She is a never smoker and has no relevant respiratory exposures.
Labs were significant for ESR of 121 mm/hr, CRP of 69 mg/L, CK of 381 U/L, aldolase of 9.1 U/L, ANA with a cytoplasmic pattern and a titer of 1:320, positive anti-PL12 antibody, and positive anti-SSA IgG antibody.
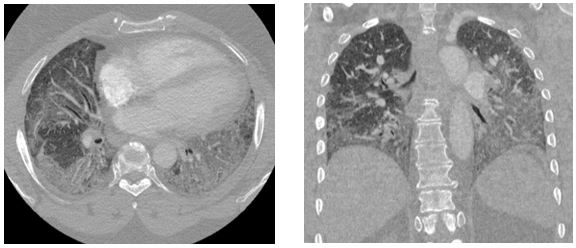
Representative images from her chest CT scan are shown below. Bronchoscopy with transbronchial biopsy and microbiological analysis were negative for evidence of infection, malignancy, and granulomatous disease.
Question
What is the most likely diagnosis?
- Scleroderma-related ILD
- Anti-synthetase syndrome
- Polymyositis
- Acute exacerbation of pulmonary sarcoidosis
- Sjogren’s syndrome
- Community acquired pneumonia
B. Anti-Synthetase Syndrome
Discussion
Anti-synthetase syndrome is an autoimmune condition characterized by antibodies directed against a number of aminoacyl tRNA synthetases and occurs in one-third of patients with polymyositis (PM) or dermatomyositis (DM).1 Although anti-synthetase syndrome is technically classified as an idiopathic inflammatory myopathy (IIM), the phenotypic behavior of anti-synthetase syndrome is much unlike PM and DM in that it commonly presents with a severe, rapidly progressive, steroid-resistant ILD.
The clinical manifestations of anti-synthetase syndrome are dependent upon the presence of specific autoantibodies directed against a number of individual amino-acyl tRNA synthetases, of which anti-Jo1 antibodies are the most frequently encountered (60-80% of cases).2 Anti-PL7 and anti-PL12 antibodies occur less frequently (5-15% of cases) but are associated with a higher incidence of ILD and worse outcomes.2 Mechanic’s hands, characterized by hyperkeratosis and scale most often of the palmar aspect of the hands, was associated with all aminoacyl tRNA synthetase antibody subtypes but occurred most frequently (56% of cases) with anti-Jo1.3,4
The diagnostic criteria for anti-synthetase syndrome were revised in 2011 by Soloman et al and require the presence of an anti-aminoacyl tRNA synthetase antibody in all cases plus (a) two major criteria, or (b) one major and two minor criteria:4
Major criteria:
- Presence of ILD
- Polymyositis or dermatomyositis by Bohan and Peter criteria
Minor criteria:
- Non-erosive arthritis
- Raynaud’s phenomenon
- Mechanic’s hands
High-resolution computed tomography (HRCT) is the imaging modality of choice for evaluation of the pulmonary parenchyma in patients suspected of having ILD. In one retrospective analysis of HRCT data, anti-synthetase syndrome presented most commonly (52% of cases) with peripheral areas of ground-glass attenuation and reticulation in a predominantly lower lobe and peribronchovascular distribution, consistent with fibrosing non-specific interstitial pneumonia (NSIP).5 Organizing pneumonia (OP) with fibrosis was the next most common HRCT pattern encountered (34% of cases) and is thought to be associated with a worse prognosis.5
Corticosteroids are the mainstay of treatment for patients with IIMs. However, anti-synthetase syndrome does not typically respond to steroid monotherapy and is associated with a relatively high rate of disease relapse with steroid tapering.4 Thus, a swift institution of multimodal treatment with adjunctive immunosuppressive agents is recommended, but there is controversy regarding which agent is preferred.
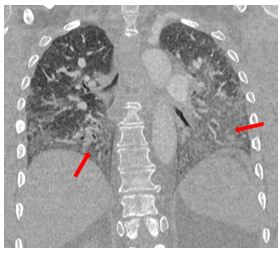
Our patient met the 2011 diagnostic criteria for anti-synthetase syndrome. She had a positive PL-12 antibody and an elevated anti-SSA 52 antibody of 104 AU/mL, but the remainder of her extended myositis antibody panel results were negative. CT imaging obtained during her hospitalization demonstrated areas of ground glass attenuation in the bilateral lower lobes (red arrows), consistent with NSIP. She experienced a rapid improvement in her dyspnea with methylprednisolone 1000 mg daily for three days. She was discharged with a one-month course of prednisone 1 mg/kg daily with close follow-up in multidisciplinary ILD clinic wherein she continued to experience improvement with the initiation of mycophenolate mofetil 1500 mg twice daily.
References
-
Dalakas MC. Inflammatory muscle diseases. N Engl J Med. 2015;372(18):1734-1747.
-
Shi J, Li S, Yang H, et al. Clinical Profiles and Prognosis of Patients with Distinct Antisynthetase Autoantibodies. J Rheumatol. 2017;44(7):1051-1057.
-
Hamaguchi Y, Fujimoto M, Matsushita T, et al. Common and Distinct Clinical Features in Adult Patients with Anti-Aminoacyl-tRNA Synthetase Antibodies: Heterogeneity within the Syndrome. Miller F, ed. PLoS ONE. 2013;8(4):e60442.
-
Witt LJ, Curran JJ, Strek ME. The Diagnosis and Treatment of Antisynthetase Syndrome. Clin Pulm Med. 2016;23(5):218-226.
-
Waseda Y, Johkoh T, Egashira R, et al. Antisynthetase syndrome: Pulmonary computed tomography findings of adult patients with antibodies to aminoacyl-tRNA synthetases. Eur J Radiol. 2016;85(8):1421-1426.


