Progressive Respiratory Failure in a Term Infant
June Hu, M.D. 1, Frances White, M.D. 2, F.S. Cole, M.D. 1, Jennifer Wambach, M.D., M.S. 1
1 Edward Mallinckrodt Department of Pediatrics,
Washington University School of Medicine, St. Louis, Missouri, United States 63110
2 Department of Pathology and Immunology,
Washington University School of Medicine, St. Louis, Missouri, United States 63110
Case
A term female infant, born of an uncomplicated pregnancy to a 20-year-old primigravida mother via cesarean section, developed respiratory distress immediately after birth requiring intubation and mechanical ventilation. Her initial chest radiograph was consistent with hyaline membrane disease and she was treated with two doses of surfactant within 48 hours after birth with transient improvement. She continued to have persistent hypoxemic respiratory failure despite escalating care which included conventional mechanical ventilation, high-frequency oscillatory ventilation and inhaled nitric oxide. On day 9 of life, computed tomography of the chest demonstrated diffuse ground glass opacities. A comprehensive infectious disease workup was negative. She was unresponsive to steroid therapy including dexamethasone (0.15 mg/kg/day for 10 days) and three courses of methylprednisolone (30 mg/kg/day for 3 days). Open lung biopsy performed at day of life 15 demonstrated interstitial lung disease with pneumocyte hyperplasia and alveolar proteinosis (figure 1). Immunohistochemical staining demonstrated aberrant surfactant expression. Electron microscopy demonstrated abnormal appearing lamellar bodies (figure 2). Genetic testing for disorders of surfactant metabolism was sent. She was treated with exogenous surfactant every 12-48 hours prior to referral for evaluation for lung transplantation.
Figure 1. Hematoxylin and eosin stained lung sections (20X) demonstrate diffuse type II pneumocyte hyperplasia (arrows), multinucleated alveolar macrophages (arrowheads), proteinaceous material in the airspaces (asterisks) and early fibrosis.
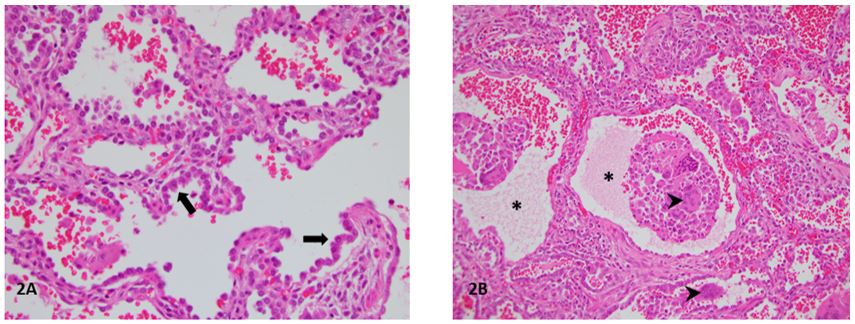
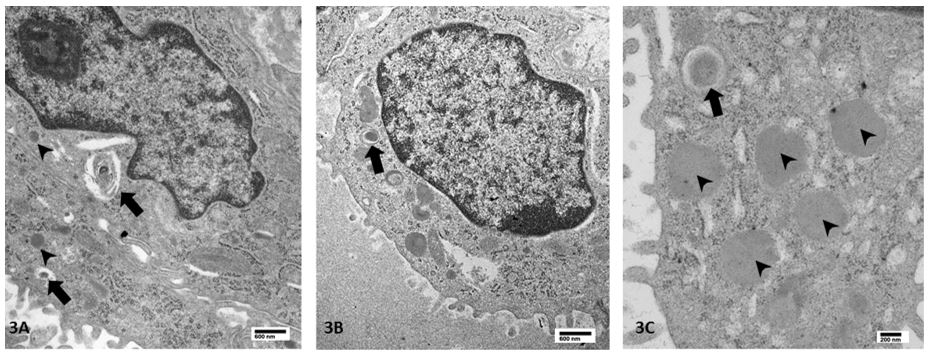
Figure 2. Electron microscopy of lung explant. 3A: Small, dense bodies (arrowheads) and few heterogeneous structures with vesicles and membranes without definite lamellar body structures (arrows). 3B: Dense body structures in which compact concentric membranes are seen (arrow). 3C: Higher power view demonstrates rare small, dense body-like structures with compact lamellar membranes at periphery (arrow), in addition to frequent small dense bodies typical of this disease process (arrowheads).
Question
Which of the following disrupts phospholipid transport into lamellar bodies where surfactant is assembled, and is the most common genetic cause of disruption of surfactant metabolism?
- Surfactant protein B deficiency
- ABCA3 deficiency
- Mutations in the surfactant protein C gene
- Mutations in NKX2-1 which encodes thyroid transcription factor 1
- None of the above
B. ABCA3 deficiency
Discussion
ATP-binding cassette transporter A3 (ABCA3) is a transmembrane protein localized to the limiting membrane of lamellar bodies (LBs) in alveolar type II cells. ABCA3 transports phospholipids, including dipalmitoylphosphatidylcholine, from the cytoplasm into LBs where surfactant is assembled.1 Rare, recessive, loss-of-function mutations in ABCA3 are the most common cause of genetic surfactant dysfunction in humans.2 Biallelic null (nonsense and frameshift) mutations in ABCA3 result in lethal neonatal respiratory failure or death without lung transplant by 1 year of age.2 The effects on protein function and disease phenotype for missense mutations, splicing variants and in-frame insertion/deletions are less predictable and range from neonatal respiratory failure to childhood interstitial lung disease.2
In vitro characterization of ABCA3 mutations suggests 2 mechanistically distinct categories: type I mutations disrupt intracellular trafficking of ABCA3 to LBs whereas type II mutations reduce ATPase activity and phospholipid transport.3 Diagnosis of ABCA3 deficiency is made through genetic testing of the patient and both parents. Additional testing that can guide diagnosis including high-resolution CT of the lungs, immunohistochemical staining demonstrating undetectable ABCA3 expression, and electron microscopy demonstrating abnormal ultrastructure of lamellar bodies. Therapies for ABCA3 deficiency are generally supportive and include optimizing oxygenation/ventilation and nutrition. Surfactant replacement, steroids, azithromycin and hydroxychloroquine have been used as medical therapies without sustained effects.4 Because mutation-specific therapies are not available, lung transplantation remains the only definitive treatment.5
Genetic sequencing for this infant revealed biallelic missense and frameshift mutations in ABCA3. Immunohistochemical staining demonstrated expression of surfactant protein B, surfactant protein C, surfactant protein D, and pro surfactant protein C while ABCA3 expression was not detected. Electron microscopy demonstrated abnormal lamellar bodies with dense eccentric cores and no normal appearing lamellar bodies (figure 2). She underwent lung transplantation at 4 months of age. Her post-operative course has been complicated by persistent weakness which prompted tracheostomy, inability to maintain adequate oral nutrition which necessitated gastrostomy tube placement and speech and motor developmental delays.
References
-
Mulugeta S, Gray JM, Notarfrancesco KL, Gonzales LW, Koval M, Feinstein SI, Ballard PL, Fisher AB, Shuman H. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J Biol Chem 2002;277(25): 22147-55.
-
Wambach JA, Casey AM, Fishman MP, Wegner DJ, Wert SE, Cole FS, et al. Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med 2014; 189(12): 1538-43.
-
Wambach JA, Yang P, Wegner DJ, Heins HB, Kaliberova LN, Kaliberov SA, Curiel DT, White FV, Hamvas A, Hackett BP, Cole FS. Functional characterization of ABCA3 mutations from infants with respiratory distress syndrome. Am J Respir Cell and Mol Biol 2016; 55(5): 716-721.
-
Kroner C, Wittmann T, Reu S, Teusch V, Klemme M, Rauch D, Hengst M, Kappler M, Cobanoglu N, Sismanlar T, Aslan AT, Campo I, Proesmans M, Schaible T, Terheggen-Lagro S, Regamey N, Eber E, Seidenberg J, SChwerk N, Aslandidis C, Lohse P, Brasch F, Zarbock R, Griese M. Lung disease caused by ABCA3 mutations. Thorax 2017; 72(3): 213-20.
-
Eldridge WB, Zhang Q, Faro A, Sweet SC, Eghtesady P, Hamvas A, Cole FS, Wambach JA. Outcomes of Lung Transplantation for Infants and Children with Genetic Disorders of Surfactant Metabolism. J Pediatr 2017; 184: 157-64.


