Never Ending Wet Cough
Paola Di Filippo
Department of Pediatrics at University of Study “G. D’Annunzio”, Chieti, Italy
Case:
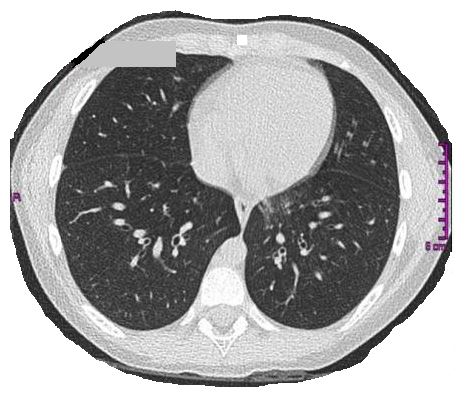
A seven-year-old girl presented to the hospital for an acute asthma exacerbation and subsequently developed respiratory failure, requiring ICU intervention. Her family history was negative for respiratory disorders and both parents were non-smokers who were in good health. She was born at term after spontaneous delivery but presented to the NICU in respiratory distress, which resolved with oxygen supplementation. Her past medical history included persistent mucous rhinitis from an early age and later, a productive cough that was unresponsive to antibiotics or inhaled steroids. Prior work-up included negative sweat chloride and CF genetic testing, negative allergy and immunology testing, and severe GERD in infancy. A representative slice of her chest CT scan is shown below.
Question:
What does the CT image demonstrate?
- Normal chest imaging
- Interstitial thickening
- Centrilobular nodules associated with some bronchiectasis in the basal segment of the lower left lobe
- Nodularity of the left lower lobe
C. Centrilobular nodules associated with some bronchiectasis in the basal segment of the lower left lobe
Discussion:
Sinus and chest CT scans of our patient revealed the “presence of pansinusitis; some nuanced centrolobular nodules associated with some bronchiectasis in the basal segment of the lower left lobe; concomitant thickening of the bronchial walls. Our patient then underwent nasal nitric oxide (nNO) testing, which was low (left: 131 ppb; right: 137 ppb). In order to evaluate ciliary motility and ultrastructure, an epithelial sample was obtained from the upper airways. Ciliary movement analysis documented poor ciliary activity with an average beat frequency of 1.7 Hz (range 0.5 - 3.5 Hz). Signs of inflammation were observed in 28% of fields and immobile cilia in 92% of fields. Ciliary ultrastructure revealed 100% pathological cilia analyzed due to an absence of internal and external dynein arms. No known genetic mutations were found, but the patient was diagnosed with Primary Ciliary Dyskinesia (PCD) based on the clinical and diagnostic features.
PCD is a clinical and genetic heterogeneous group of respiratory ciliopathies, with reduced mucociliary clearance of the airways. The prevalence is approximately 1 per 10,000 births. Although PCD is a rare disease, its main presenting symptoms are common in the pediatric population. Clinical manifestations include respiratory distress in at least 80% of full-term newborns. Patients also present with chronic upper and lower airway disease (year-round daily nasal congestion, chronic cough, otitis media with effusion and recurrent lower respiratory infections), left-right laterality defects and infertility. A persistent, year-round, daily wet cough and nasal congestion from early infancy are documented in nearly 100% of PCD patients. Respiratory symptoms do not completely resolve with antibiotic therapy or seasonal changes. By preschool age, up to 80% of PCD patients have recurrent lower respiratory tract infections, but frequent treatments with antibiotics for rhinorrhea or otitis media may mask this. Fifty percent of children will have bronchiectasis by 8 years of age. Situs inversus is present in approximately 50% of patients and heterotaxy defects are attributed to the loss of function of nodal cilia during embryogenesis.
American Thoracic Society (ATS) guidelines suggest that diagnostic testing is mandatory in patients with at least two of four key features: 1) year-round, daily, wet cough; 2) year-round, daily, nonseasonal rhinosinusitis; 3) neonatal respiratory distress syndrome in term newborns; 4) laterality defects in the absence of a single definitive test for PCD, the ATS recommends that a combination of nasal nitric oxide (nNO), transmission electron microscopy (TEM) and genetic testing are required for a definitive diagnosis. Diagnostic criteria differ in Europe, where diagnosis is currently based on a combination of high speed videomicroscopy analysis (HVMA), TEM, genetic test results and abnormally low nNO. Screening for PCD by measuring nNO during a velum closure maneuver is accurate with good sensitivity and specificity and it is relatively easy to perform in non-cooperative children. TEM can show most defects associated with pathological cilia immobility, but up to 30% of patients have a normal ciliary ultrastructure; therefore, normal ciliary TEM cannot rule out a PCD diagnosis. Furthermore, inflammation and infections can alter the normal 9+2 cilia ultrastructural arrangement. Therefore, it can be difficult to differentiate acquired defects from PCD. Several studies have suggested that genetic testing identifies the gene in ~65% of cases.
ATS PCD guidelines suggest:
- Using an extended genetic panel (testing > 12 genes) as a diagnostic test over the reference standard of TEM and/or standard genetic panel (≤12 genes) testing;
- Using nasal nitric oxide testing in patients over the age of five, who have tested negative for cystic fibrosis (one-third of CF patients can have nNO values below PCD diagnostic cutoffs);
- Not using ciliary beat frequency analysis by digital high-speed videomicroscopy as a replacement diagnostic test over the reference standard.
To date, there are no evidence-based treatment guidelines informed by randomized clinical trials in patients with PCD. Thus, physicians treating PCD adapt therapeutic approaches used for other chronic respiratory diseases, such as cystic fibrosis and non-CF bronchiectasis.
References:
-
Fitzgerald DA, Shapiro AJ. When to suspect primary ciliary dyskinesia in children. Paediatr Respir Rev 2016; 18: 3-7.
-
Mullowney T, Manson D, Kim R et al. Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics 2014; 134: 1160-6.
-
Shapiro, Zariwala MA, Ferkol T et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Ped Pulmon 2016; 51:115-132.
-
Collins SA, Gove K, Walker W et al. N asal nitric oxide screening for primary ciliary dyskinesia: a systematic review and meta-analysis. Eur Respir J 2014; 44:1589-1599.
-
Lucas JS, Barbato A, Collins SA, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J 2017; 49.
-
Shapiro AJ, Davis SD, Polineni D et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Journal of Respiratory and Critical Care Medicine 2018; 197: 24-40.


